In this post I am laying out a framework to think about the space between targeted and untargeted approaches, all of which logically carries the name Semi-targeted.
Last week I had a friend tell me he was doing a “semi-targeted” analysis by retrieving only peaks that he knew he could name. He still couldn’t quantitate them but at least he thought he knew what they were. He was justifiably proud to be doing a higher grade of metabolomics work than he had when he first started. I was too.
Currently, Metabolomics is basically divided into two camps, Targeted metabolomics and untargeted (sometimes called unbiased) metabolomics. The Targeted work is generally based on clinical style analytical measurements of known compounds that are quantitative, with the implicit need to compare or normalize different samples against one another. On the other hand, untargeted metabolomics experiments represent the need to make metabolomics a true omics science in which one measures everything, and tries to determine which peaks have a possible statistical relationship with different experimental subgroups. Untargeted metabolomics initially started out as biomarker discovery on steroids, pre-Bonferroni, and it was not unusual for an untargeted analysis to consider every one of the peaks that were seen in the mass spectrum. This sometimes meant including 30,000 to 40,000 peaks and was called the Complete Reporting Analytical Protocol.
The reality is that these two techniques represent ideals that most researchers do not conform to. Most people, like my friend, do not do pure targeted or untargeted work but rather work somewhere between these two goals. Instead they take shortcuts or make enhancements according to their abilities or needs to balance the quantity of data they can get while still guarding data quality. After all, generating the dataset is the easy part, maybe a day or two, while the analysis of the data may take months, so why waste your time with bad data.
Below I am proposing what I believe is a reasonable framework for thinking about semi-targeted approaches.
I am assuming that there are three main ways that most researchers try to enhance their data and correct or reduce error:
- The first is by correctly identifying the peaks to compounds,
- The second is by correcting (normalizing) the dataset against sample-to-sample or preparation incurred variations, and
- The third is by correcting for in-source errors and inefficiencies.
If each semi-targeted technique is scored on each of these aspects on a zero to ten scale (where zero is no effect, and ten is a complete solution) then any Semi-targeted analysis protocol can be placed somewhere in this three-dimensional space.
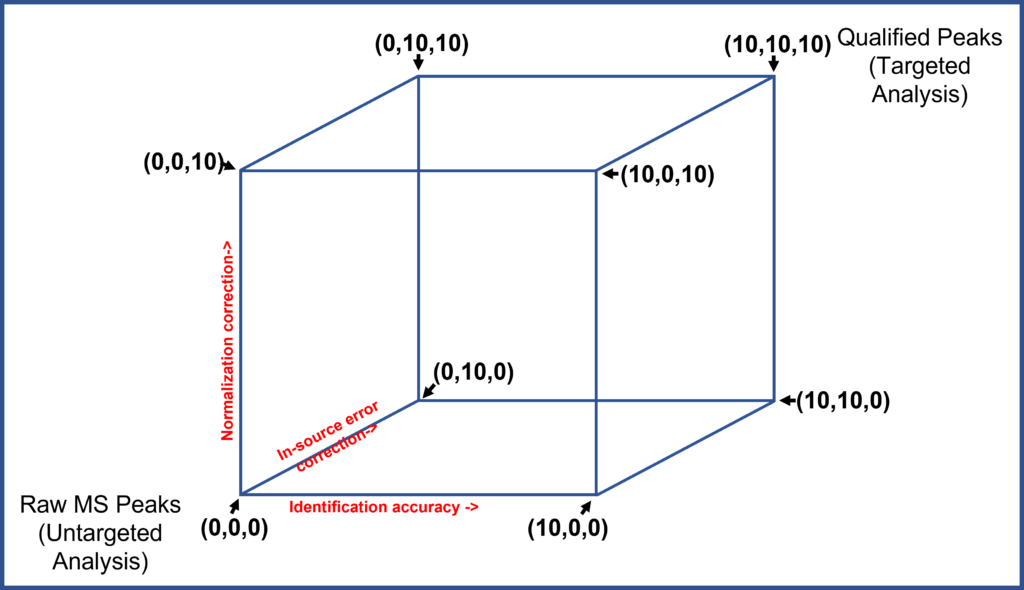 Example: My friend knows a lot of compounds and by using only peaks that he has positively identified let’s say he can get a 6 out of 10 for ability to identify all his peaks. And let’s say he has no ability to correct for source errors, and no normalization scheme in his normal workflow, so his method rests at coordinates (6, 0, 0) in this space.
Example: My friend knows a lot of compounds and by using only peaks that he has positively identified let’s say he can get a 6 out of 10 for ability to identify all his peaks. And let’s say he has no ability to correct for source errors, and no normalization scheme in his normal workflow, so his method rests at coordinates (6, 0, 0) in this space.
If he introduced a sample-to-sample normalization routine based on some internal standards, say a 3 of 10 score in normalization, then his protocol would be a semi-targeted protocol with a (6, 0, 3). If his routine used more internal standards and a standard sample to correct for source errors, this may give him a semi-targeted profile of (6, 2, 3).
This seems pretty straight-forward. There are a number of well-known techniques for correcting each of these error axes. If each of them could be assigned a score then we could likely put all of the techniques that researchers are using and develop a picture of where the world of semi-targeted analyses are today. It might surprise us. It might be fun, and it would surely lead to better metabolomics tomorrow.
Please send me a quick note (chris@iroatech.com) about whether your laboratory workflow is targeted, semi-targeted, or non-targeted and what you think of this. Additional aspects in need of correction that you can think of? Techniques that you would recommend for correcting any of these error axes?





